研究重点解析:
1. 在慢性HF患者中,植入LVAD后心肌恢复且MPC表达增加
2. MPC缺失足以诱导心肌肥大和HF
3. MPC过表达则减弱药物诱导的肥大
4. 线粒体丙酮酸转运失衡是诱导心肌细胞肥大的充分必要条件
5. 抑制MCT4可以减轻细胞和小鼠的心肌肥大
心力衰竭(heart failure,HF)是一种复杂的慢性心脏病,其特征是心脏的泵血或血液再灌注能力降低,已成为全球死亡的主要原因。HF的发生通常以病理性心肌肥大为先导征兆。晚期HF患者的治疗主要是侵入性手术,例如心脏移植和左心室辅助装置(LVAD)植入,以此通过机械支持恢复足够的血流量和血液动力学。然而植入LVAD后一部分患者有效而另一部分患者却效果不佳,其原因尚不清楚。来自美国犹他大学Stavros G. Drakos和Jared Rutter团队利用RNA测序、葡萄糖示踪及代谢组学等技术发现,在慢性HF患者中,植入LVAD后心肌恢复伴随着MPC(线粒体丙酮酸载体)表达增加,同时揭示了丙酮酸-乳酸轴的改变是心脏肥大和心力衰竭的早期特征,抑制MCT4可以改善HF,相关研究成果发表于《Cell Metabolism》。
线粒体丙酮酸载体(MPC)是MPC1和MPC2的异二聚体复合体,它将糖酵解的产物丙酮酸运输到线粒体,在线粒体中通过丙酮酸脱氢酶(PDH)和三羧酸(TCA)循环完全氧化。MPC亚基在正常成人心脏中高表达,可能是决定丙酮酸氧化或胞质转化为乳酸的关键节点。
一. MPC1缺失促进心肌肥大导致HF
晚期慢性心力衰竭患者植入LVAD后,患者被分为有应答者和无应答者(应答者组定义为LVEF增加大于50%,LVEDD 小于6.0 cm,无响应组定义为LVEF增加小于50%,无论LVEDD是否改变)。植入LVAD后,与无应答者不同,有应答组的心肌收缩功能指标(LVEF)显著增加,而心脏大小指标(LVEDD)显著减少。收集并分析两组HF患者在植入LVAD前和植入LVAD后的左心室组织,以明确应答者心力衰竭恢复的机制。MPC1丰度检测结果显示,与供体对照组相比,在植入LVAD前,应答组和非应答组的心肌组织中MPC1均降低;而植入LVAD后,心肌组织中MPC1在应答组中增加,但在无应答组的MPC1丰度不变。检测丙酮酸脱氢酶(PDH)的磷酸化程度(PDH被磷酸化后失活,去磷酸化后激活),结果显示,在应答组中,与供体对照组相比,在植入LVAD 前显著升高,在植入LVAD 后降低并接近对照水平;而在无应答组植入LVAD前和后,与供体对照组相比,PDH磷酸化程度均增加,导致PDH失活,提示丙酮酸氧化受到抑制,可能存在一种潜在的代谢病因,以线粒体丙酮酸代谢缺陷为中心导致心衰。
由于MPC在HF患者中较低,而在应答者植入LVAD后增加,那么MPC表达的改变是否对HF和康复有因果关系?为了确定MPC缺失是否足以在体内诱导HF,构建心脏特异性和他莫昔芬诱导的MPC1敲除小鼠(MPC1CKO),MPC1敲除小鼠表现为心脏特异性和他莫昔芬诱导的MPC1和MPC2蛋白的缺失。在他莫昔芬治疗1周后,对这些小鼠进行连续的超声心动图检查,结果显示,注射他莫昔芬1周后MPC1敲除小鼠与野生型(WT)幼崽无区别;而在诱导9周后(9 wpi), MPC1敲除小鼠表现出扩张性心肌病,到16 wpi时进展为明显的心衰。组织切片结果表明,与WT小鼠相比,MPC1敲除小鼠的心脏横断面积增加。从13wpi开始左室重量增加,也提示MPC1敲除小鼠的心肌肥大。在13 wpi后,MPC1敲除小鼠体重显著下降,之后开始死亡,并在18 wpi时全部死亡。综上所述,小鼠中MPC1缺失足以引起心肌肥大和心衰(图1)。
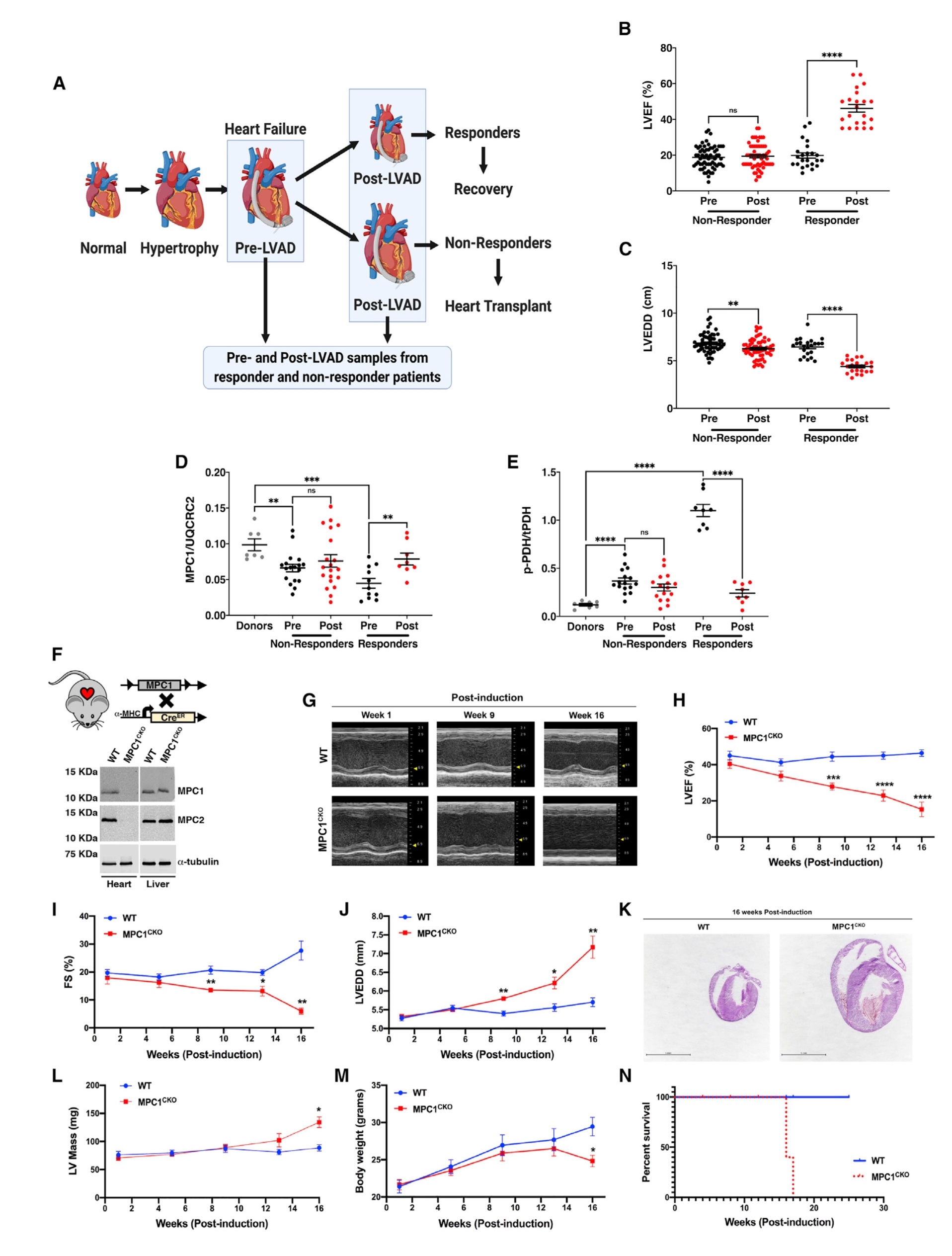
图1. MPC1缺乏足以促进心肌肥大并导致心力衰竭
二. MPC缺失导致早期肥大和最终线粒体衰竭
将8 wpi(肥大前)和16 wpi(心衰后)的对照组和MPC1敲除小鼠的心脏组织进行RNA测序,并对差异上调基因进行GO分析,结果显示,与WT小鼠相比,MPC1敲除小鼠中参与心肌细胞发育、胶原组织、细胞分裂和损伤反应的基因表达均上调,在8周上调明显,并在16周进一步加强,提示肥大前基因调控程序的启动。当检查单个基因时,与WT小鼠相比,MPC1敲除小鼠中与心肌肥大和糖酵解相关的基因表达显著升高,与TCA循环酶相关基因表达降低;还发现,在8 wpi和16 wpi时,丙酮酸代谢相关基因调控失调。MPC1敲除小鼠的左室组织透镜(TEM)结果显示,在8 wpi时,线粒体结构正常;而在16 wpi时,线粒体嵴不规则,染色强度较高。综上所述,虽然MPC的缺失引起了代谢基因表达的早期代偿性变化,但最终还是导致线粒体功能紊乱,发生HF和死亡(图2)。
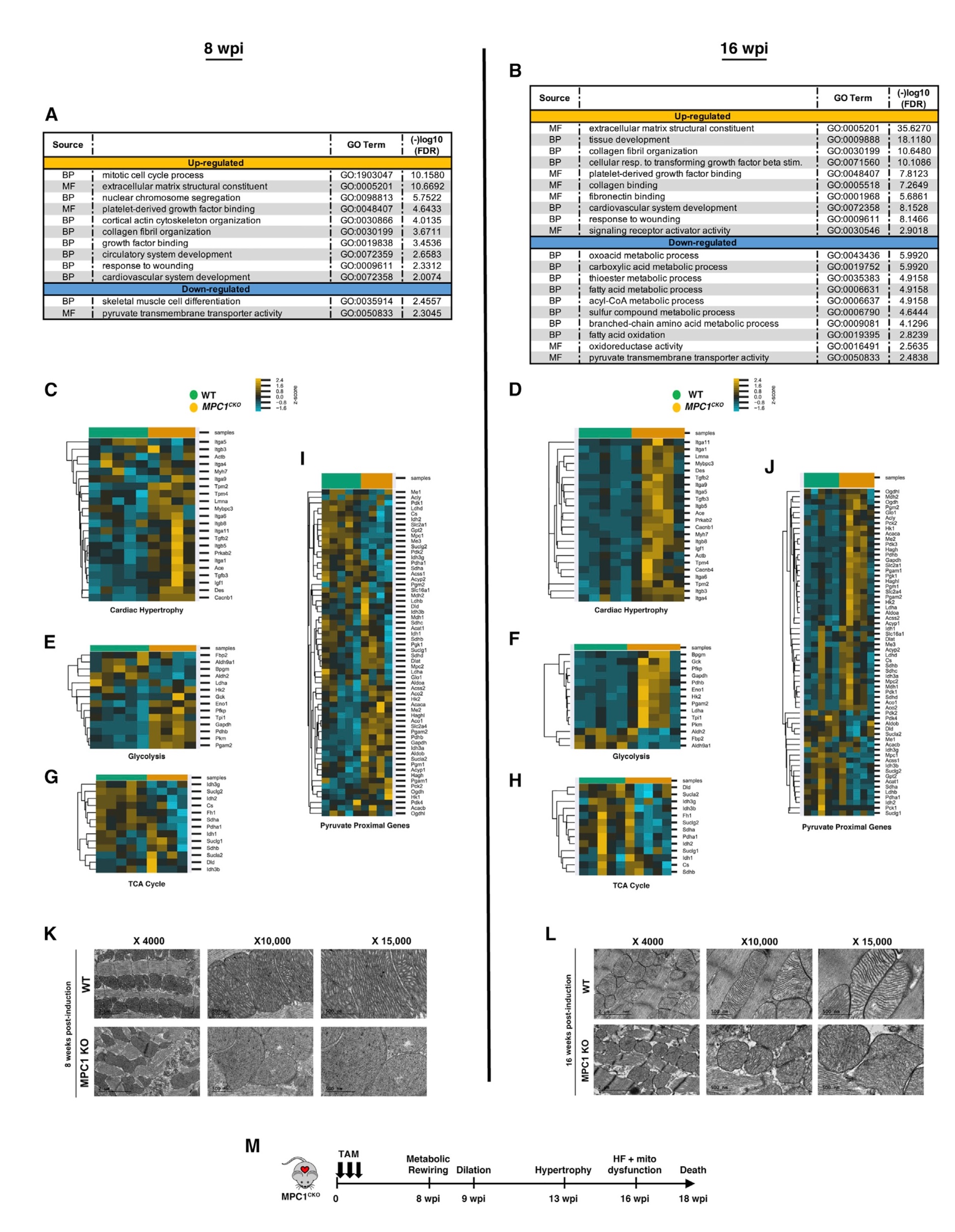
图2. MPC丢失是一种心衰的早期损伤
三. 线粒体丙酮酸转运失衡是诱导心肌细胞肥大的必要条件
如果MPC活性的丧失是心肌细胞肥大的早期损伤诱因,那么MPC的过表达可能会导致对诱导肥大药物的抵抗。用MPC过表达载体(MPC OE)或空白载体(EV)转染H9c2细胞(它可以分化成具有许多心肌细胞特征的细胞),同时使用肥大诱导药物苯肾上腺素(PE)、异丙肾上腺素(ISO)和血管紧张素(ATII)以及MPC抑制剂UK-5099 (UK)处理MPC OE和EV细胞,结果显示,MPC OE可以减轻所有四种药物诱导的细胞肥大。由于MPC是一种蛋白质复合物,它将胞质丙酮酸转运到线粒体基质中,并没有内在的酶活性。因此,切断这种联系就足以促进肥大。为了证实这一点,使用葡萄糖示踪技术分析上述相同实验条件下碳水化合物代谢通量的变化,结果显示,在UK、MPC1 KD(敲低,基因干扰)和PE处理的H9c2细胞中,细胞内和细胞外同位素标记的乳酸均增加,而同位素标记的柠檬酸在TCA循环中下降。这些代谢扰动不只在MPC抑制或敲低后发生,也在PE处理后发生。且MPC OE处理后,则逆转了 PE 诱导的 TCA 循环中间体的降低,同时防止了细胞肥大,这进一步支持了这种代谢转移对细胞肥大的必要性(图3)。
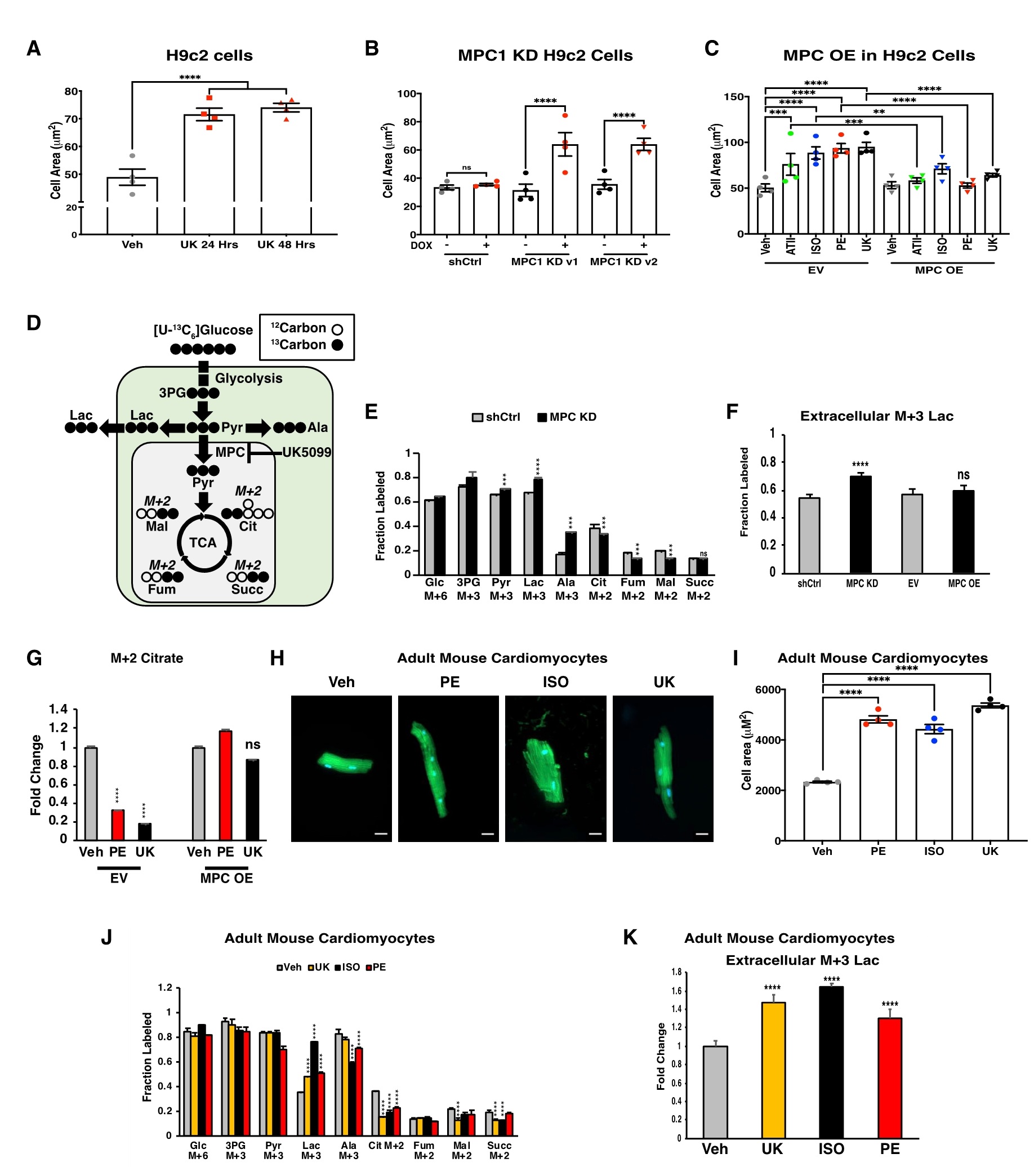
图3. 线粒体丙酮酸转运的丧失是诱导培养心肌细胞肥大的必要条件
四. 线粒体丙酮酸转运失衡是诱导心肌细胞肥大的充分条件
为了更好地了解代谢通量变化、肥大和 HF 之间的关系,从MPC1敲除小鼠中分离培养 ACMs(成年小鼠心肌细胞)。在心肌结构或功能显著异常前(12 wpi)和心衰发作后(16 wpi)分别分离心肌细胞,并进行葡萄糖示踪实验。结果表明,与MPC活性丧失一致,在12 wpi时,MPC1敲除的ACMs细胞中,标记的乳酸增加,柠檬酸减少,而TCA循环中间体富马酸和琥珀酸的标记不受影响。同时,还发现标记的丙氨酸水平正常,它的胞质生成和线粒体输入可以作为MPC独立的替代机制,通过这种机制,糖酵解的碳得以供给TCA循环。而在16 wpi时,进入TCA循环的标记消失,丙氨酸标记减少,乳酸输出进一步增加。这表明,线粒体丙酮酸摄取的减少足以在体外引起心肌细胞肥大,线粒体丙酮酸转运失衡是诱导心肌细胞肥大的充分条件(图4)。
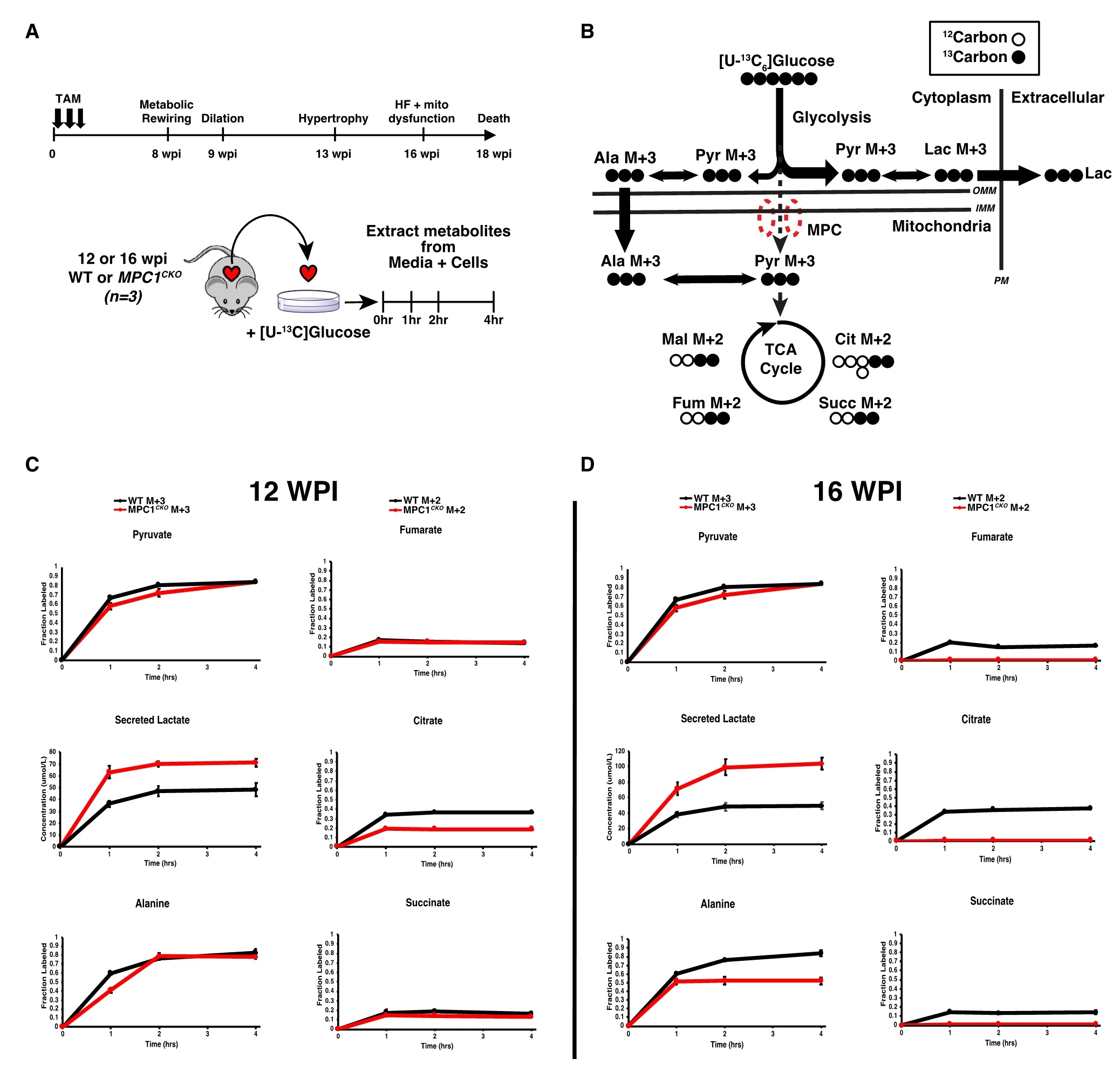
图4. MPC1CKO小鼠肥大和HF前糖酵解通量的动态变化
五. 抑制MCT4可预防和逆转心肌细胞肥大
已有文献报道单羧酸转运体(MCT)家族的成员MCT4在肥大和HF中高表达,从而将乳酸从细胞中排出。作者开发一种MCT4抑制剂VB124,它可以特异性地抑制乳酸流出,从而将糖酵解碳通量转向线粒体丙酮酸氧化。假设抑制MCT4阻断了肥大心肌细胞的乳酸外流,导致糖酵解碳通量转向线粒体丙酮酸氧化,进而逆转心肌肥大。为了验证这一假设,将PE和VB124同时作用于H9c2细胞48h,结果表明,抑制MCT4可以阻止PE诱导的细胞肥大。为了确定MCT4抑制是否不仅能阻止PE诱导的细胞肥大,而且还能逆转肥大,我们用PE预处理H9c2细胞48小时,然后再联合PE和VB124处理48小时,结果表明,MCT4抑制完全逆转了这些细胞的肥大表型。用PE和ISO处理ACMs,同样发现VB124阻止了ACMs细胞的肥大表型。抑制MCT4后的逆转效应,以及MPC过表达的抗肥大效应,均体现了心肌细胞线粒体丙酮酸代谢的重要性,这与植入LVAD的患者中观察到的MPC丰度增加一致。葡萄糖示踪实验也表明,VB124 可防止在 H9c2 和 ACM 细胞中用 PE 或 ISO 处理引起的代谢异常,VB124 降低了向乳酸的葡萄糖通量,并增加了进入柠檬酸盐和 TCA 循环的葡萄糖通量。在经PE预处理、随后经VB124处理的细胞中也发现了TCA循环通量的增加。综上所述,抑制MCT4可以逆转药物诱导的心肌细胞肥大(图5)。
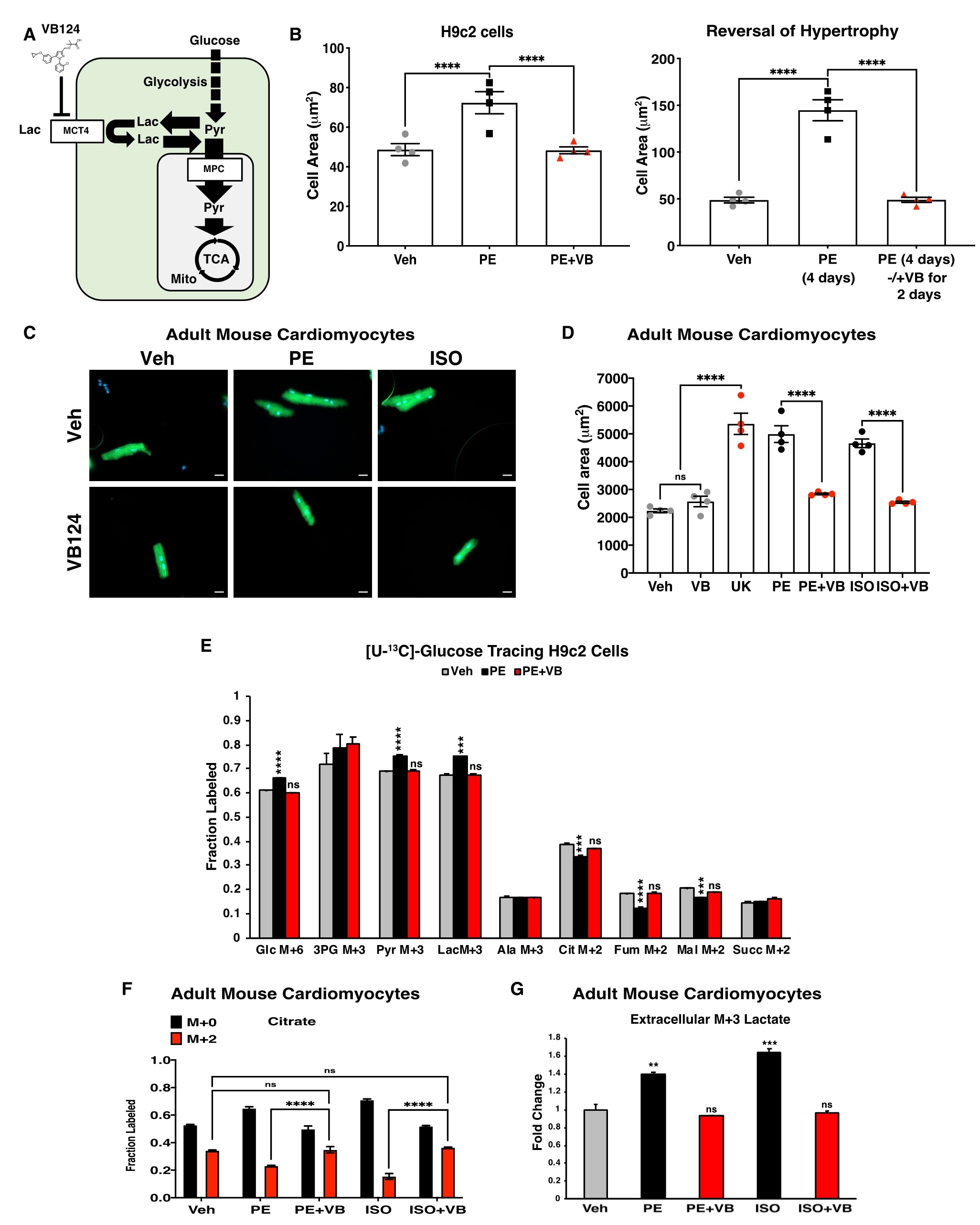
图5. 抑制MCT4可预防和逆转心肌细胞肥大
六. 维持线粒体丙酮酸可防止心肌细胞肥大
构建稳定表达线粒体特异性标签(Mito-tag)的H9c2细胞,该标签能够快速分离和提取线粒体,这是量化线粒体内代谢物改变的必要条件。检测到心肌肥大药物刺激导致稳态丙酮酸水平下降,而MCT4抑制又恢复了稳态丙酮酸水平。然而,在线粒体中未检测到乳酸,这与乳酸主要是一种细胞质代谢物的观点一致。线粒体丙酮酸含量的这种模式与全细胞提取物中趋势相反,在全细胞提取物中,UK、PE和ISO刺激导致丙酮酸和乳酸的增加,而MCT4抑制则逆转了这种情况。为了确定这种丙酮酸可能是糖酵解的来源,我们将Mito-tag标签方法与葡萄糖示踪组合,并观察到丙酮酸同位素在未处理的细胞中呈现高丰度,而在UK、PE和ISO处理中未检测到,且在MCT4抑制时再次出现。与之前的实验一致,任何情况下均未检测到标记的乳酸,这表明糖酵解产生的丙酮酸而非乳酸进入线粒体并为这些细胞的TCA循环提供燃料。同时对这些Mito-tag细胞进行不同时间梯度的葡萄糖示踪实验,再次观察到VB124处理后糖酵解进入TCA循环的通量增加。除了丙酮酸-乳酸轴的变化,还观察到在PE、ISO和UK处理的细胞中,线粒体中稳态谷氨酸增加,天冬氨酸减少,且MCT4抑制后,线粒体天冬氨酸和谷氨酸水平则完全逆转。同时也在全细胞裂解物中获得相反趋势,进一步强调了心肌细胞中分室化线粒体代谢的重要性。
糖酵解通量增加和细胞呼吸下降是心肌细胞肥大的常见现象。线粒体膜电位是ATP产生和蛋白质输入所必需的,是线粒体整体健康的可靠标志。与此一致, ISO处理后,成年心肌细胞线粒体膜电位下降,而VB124处理完全逆转了成年心肌细胞的膜电位。而且这些细胞在ISO处理后活性氧(ROS)升高,而VB124处理后发生逆转。综上所述,MCT4抑制可减轻和逆转心肌细胞肥大,使线粒体氧化代谢正常化(图6)。
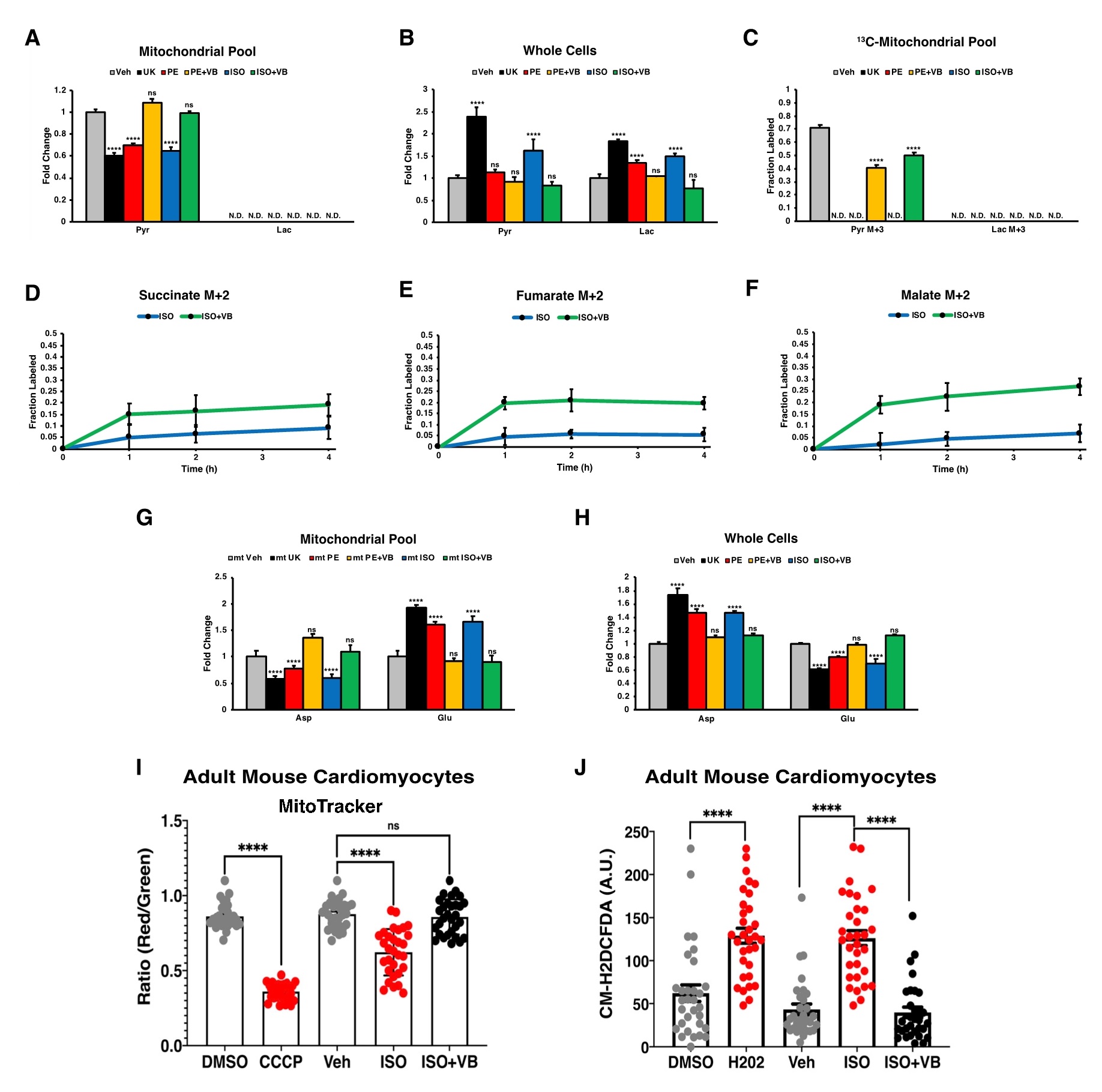
图6. MCT4抑制补充线粒体丙酮酸通量,提高线粒体膜电位,减轻细胞溶质ROS
七. 抑制MCT4可减轻ISO诱导的小鼠心肌肥大
考虑到抑制MCT4逆转了PE、ISO和UK体外治疗后的心肌细胞肥大,于是在心肌肥大的小鼠模型上再次验证。通过外科手术将小鼠植入渗透微型泵,连续输送Veh或ISO以诱导小鼠肥大,同时通过每日小鼠灌胃VB124或Veh,持续4周。在4周结束时对这些小鼠进行了超声心动图检查,并在死后收集心脏样本。与细胞结果一致,注射ISO引起心脏体重比和LVEDD显著增加,抑制MCT4时,则发生逆转。
从ISO 和 VB124 处理的小鼠中分离出的整个心脏样本进行代谢组学检测,层次聚类结果显示了ISO 处理导致的代谢变化,而 VB124 处理则逆转了这些变化,且主成分(PCA)分析结果显示,抑制MCT4足以使经过ISO处理的小鼠心脏的代谢谱恢复到与未经处理的对照组同等的水平。在ISO诱导的肥大中,糖酵解中间体增加,再用VB124处理后,则恢复到对照水平。心肌左室组织透镜分析显示,ISO处理导致线粒体结构畸变,而VB124处理改善了线粒体结构畸变,而VB124处理后则恢复。综上所述,抑制MCT4逆转了ISO诱导的心肌肥大的多种病理特征,包括代谢紊乱和线粒体结构损伤(图7)。
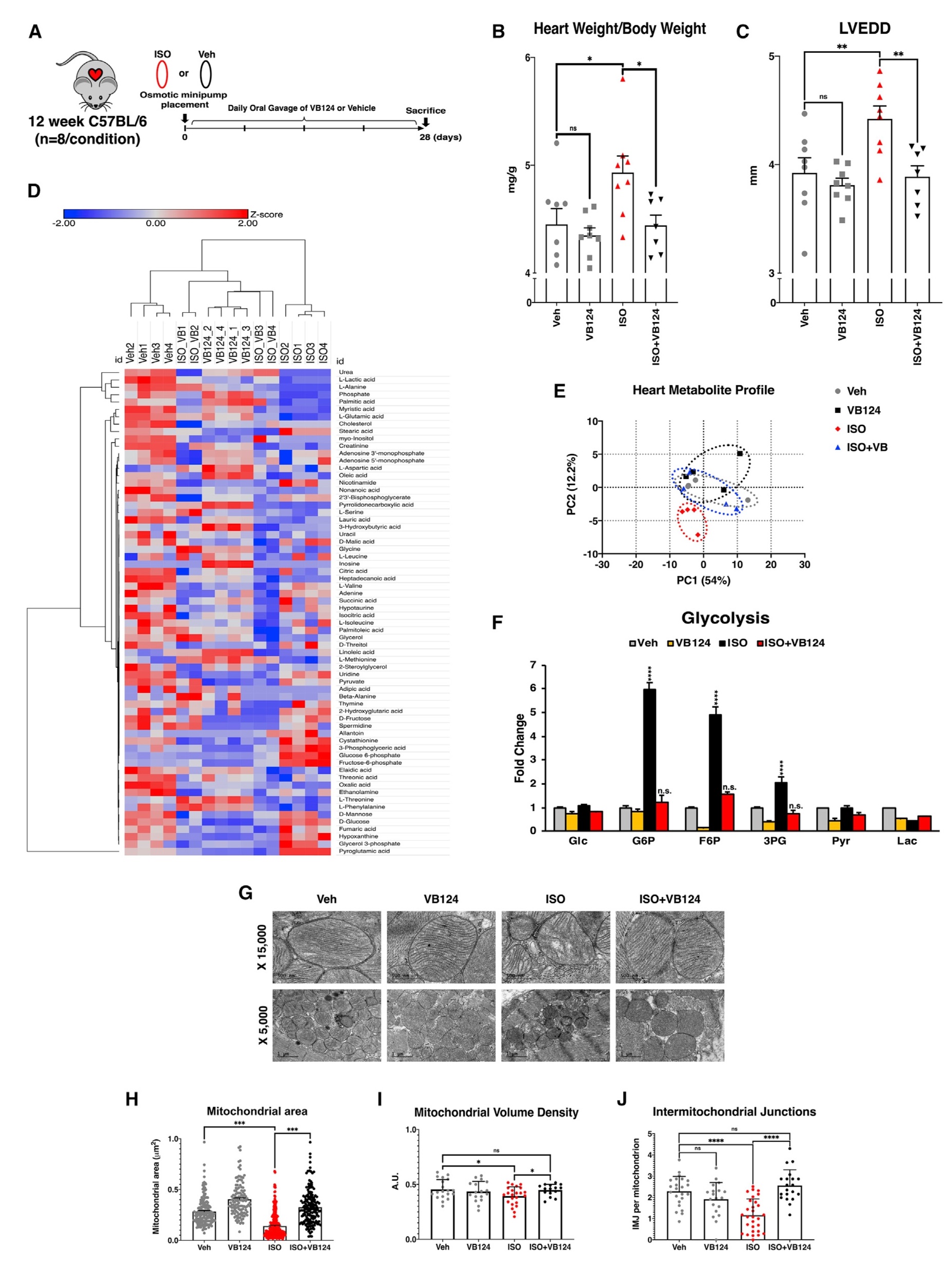
图7. MCT4抑制可防止小鼠心肌肥大
小结:
本研究发现,在植入LVAD的慢性心衰患者中,心脏恢复与心脏组织中MPC丰度的增加相关。相反,成年小鼠MPC基因缺失足以诱发心肌肥大、慢性心衰和死亡。此外,在多个体外心肌细胞模型中,MPC缺失足以诱导心肌肥大,而MPC过表达可减轻药物诱导的心肌肥大。研究还发现乳酸输出剂MCT4的活性在代谢上抑制MPC,是心肌肥大所必需的。本研究还开发了一种新型、有效的MCT4抑制剂,可以预防和逆转细胞和小鼠的心肌肥大。通过葡萄糖示踪实验和代谢组学的结合,我们发现线粒体丙酮酸氧化减少与心肌细胞肥大紧密相关。总之,本研究揭示了丙酮酸-乳酸代谢轴是心肌细胞生长和功能的一个关键调控节点,为心肌肥大疾病的有效治疗和开发高效的MCT4抑制剂提供新策略。
绘谱帮你测:
通过葡萄糖示踪实验和代谢组学的结合,上述研究发现线粒体代谢改变与心肌细胞肥大表型紧密相关。质谱代谢组学可对生物样品中的数千个小分子进行全面、系统的分析,可以灵敏捕捉到机体代谢的细微变化,是寻找线粒体相关疾病的理想生物标志物或药物靶标的最新技术。代谢组学可用于分析线粒体功能障碍的众多上下游效应,包括能量代谢、氧化应激、NAD/NADH氧化还原失衡和能量缺乏在机体系统中的变化。文中涉及的线粒体代谢及其所有相关小分子代谢物检测,麦特绘谱均可提供全套解决方案!欢迎扫码联系我们取得详细资料。
参考文献:
The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metabolism. 2020. https://doi.org/10.1016/j.cmet.2020.12.003
原文阅读,请长按识别下方二维码

精彩推荐:
1. Circulation | 短链脂肪酸–丙酸盐预防高血压心血管损害
2. Cell︱菌群代谢物-苯乙酰谷氨酰胺-心血管疾病又一推手
3. 综述 | Nature子刊:肠道菌群,如此“关”心(完整版)
4.Nature Metabolism | 为乳酸正名:能量代谢中的丑小鸭
